A Doença de Huntington (DH) é uma doença neurológica degenerativa, hereditária e rara, que geralmente se manifesta na idade adulta. É caracterizada pela degeneração gradual de partes específicas dos gânglios basais, que são conjuntos de células nervosas responsáveis pela coordenação de movimentos. As suas manifestações são motoras, psiquiátricas e cognitivas.
Sinais e sintomas da Doença de Huntington
Dentre os sintomas motores iniciais, estão movimentos involuntários esporádicos, especialmente de músculos faciais e dos dedos das mãos e pés, que, ao longo do tempo, se estendem para outros músculos. Com a evolução da doença, atos como caminhar, falar e comer são prejudicados, tornando-se lentos e descoordenados.
Outro sintoma de DH é a deterioração mental, que evolui de forma gradual. Ela impacta as funções cognitivas, incluindo perda de memória e alterações de humor, como apatia, impaciência e nervosismo.
Alteração genética
A DH possui etiologia genética, com herança dominante, o que significa que apenas uma variação genética (ou seja, “mutação”), herdada de um dos pais, é suficiente para a manifestação da doença.
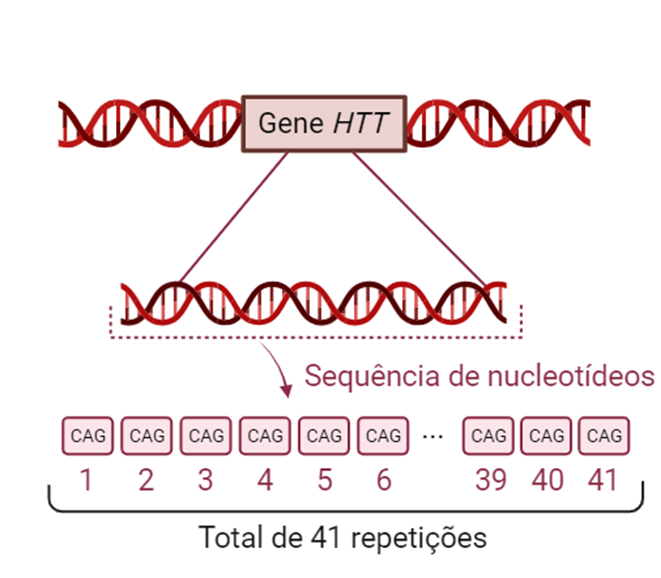
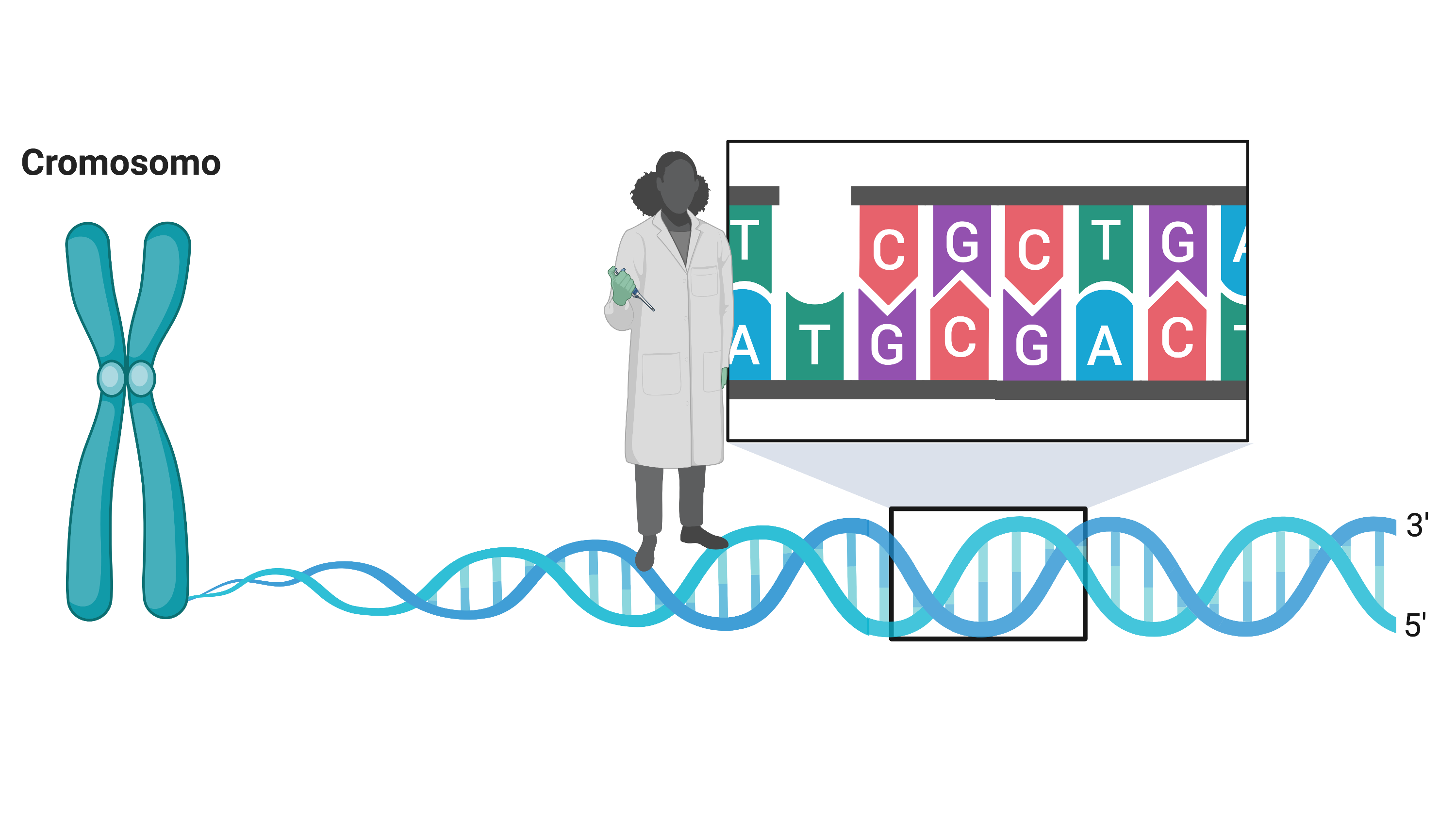
Essa variante ocorre no gene HTT, localizado no cromossomo 4, que codifica a huntingtina, proteína com papel importante relacionado ao funcionamento neuronal. A mutação causadora é caracterizada pela expansão da repetição da sequência dos nucleotídeos CAG (Citosina – Adenina – Guanina) em uma parte específica do gene. Vale citar que esse tipo mutação, caracterizada pela repetição de pequena sequência do DNA, é chamada de STR, abreviação do termo em inglês “Short Tandem Repeats”.
Diagnóstico da Doença de Huntington
O diagnóstico é realizado por meio de histórico familiar, avaliação clínica, neuroimagem e teste genético. Com relação ao último, é utilizada técnica que permite a identificação da quantidade das repetições da sequência CAG que estão presentes no gene HTT. Assim, a doença é confirmada a partir da ocorrência de 40 ou mais repetições.
Figura 1: repetição da sequência CAG no gene HTT. A quantidade dessa repetição determina a ocorrência da Doença de Huntington.
Tratamento e aconselhamento genético
É essencial que seja realizado antes, durante e após o diagnóstico o aconselhamento genético. Ele visa instruir o paciente e seus familiares sobre os riscos relacionados ao prognóstico da DH e da possibilidade de transmissão da variante no gente HTT para as gerações subsequentes. Também é muito importante para o planejamento familiar, considerando os descendentes do indivíduo com a doença, que também podem desenvolver HD caso tenham herdado a mutação.
Não existe, atualmente, cura para a DH. O seu tratamento é de suporte, ajudando a amenizar os sintomas. Dessa forma, o aconselhamento genético adequado permite que as famílias lidem melhor com a situação e tomem decisões assertivas, munidas de todas as informações necessárias.
A epilepsia é uma doença neurológica crônica, caracterizada pela predisposição do cérebro em gerar crises epilépticas de forma espontânea e recorrente. Afetando aproximadamente 1,5% da população global, essa condição pode ocorrer em indivíduos de qualquer idade, gênero, etnia ou classe social, acarretando consequências neurobiológicas, cognitivas e psicossociais.
Epilepsia genética
Existem várias causas possíveis para a epilepsia, sendo uma delas a origem genética. Em tais casos, a doença é resultante de uma mutação genética conhecida ou presumida, que tem as convulsões como um sintoma central.
É crucial esclarecer que “genético” não deve ser confundido com “hereditário”, uma vez que muitas das mutações genéticas associadas à epilepsia são do tipo “de novo” – ou seja, surgem no próprio indivíduo e não são herdadas de seus genitores. No entanto, esse indivíduo com a mutação pode transmitir o gene mutado para seus descendentes.
Diagnóstico da epilepsia
Baseado em avaliação clínica, o diagnóstico da epilepsia é feito com apoio de exames de imagem, como ressonância magnética e exames de eletroencefalografia, além da análise do histórico familiar. Em relação às epilepsias de origem genética, podem ser realizados exames específicos, como painéis de genes associados à epilepsia e sequenciamento do exoma completo.
Uma vez confirmado o diagnóstico, o tratamento deve ser iniciado rapidamente, podendo envolver tanto o uso de medicamentos anticonvulsivantes (com sucesso em até 70% dos casos) como terapias complementares, como dieta cetogênica, neurocirurgia e a implantação de um estimulador do nervo vago (VNS). Também existem outras opções que contemplam 30% das pessoas com epilepsia que não respondem ao tratamento com dois ou mais medicamentos combinados, necessitando de abordagens adicionais.
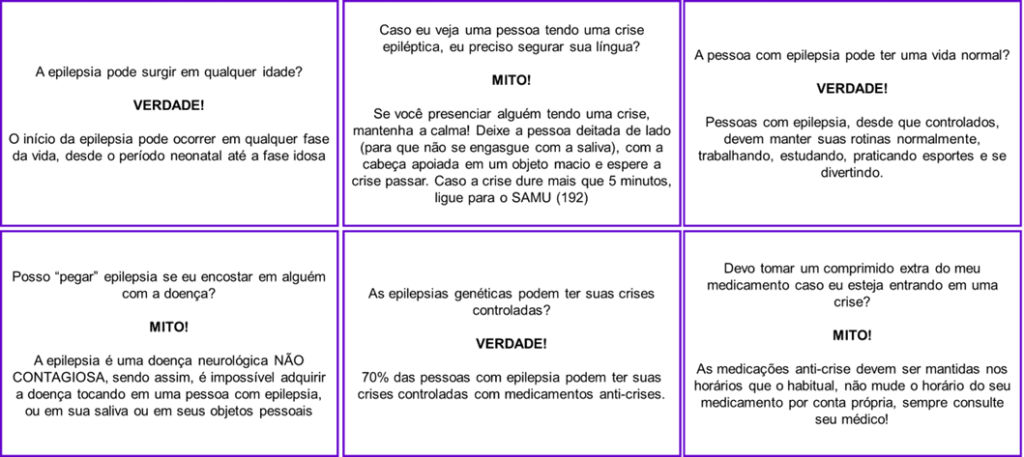
Além dos desafios médicos, as pessoas com epilepsia frequentemente enfrentam estigmas e preconceitos devido à falta de informação na sociedade. Portanto, é fundamental promover a conscientização, esclarecendo mitos e verdades relacionados à doença.
Estima-se que 80% das doenças raras possuam origem genética e cerca de 50% dos portadores não tenham um diagnóstico molecular definido. Com a descoberta da sequência completa de DNA do genoma humano em 2004, foi possível detalhar a estrutura e o modo em que os genes estão dispostos ao longo dos nossos cromossomos.
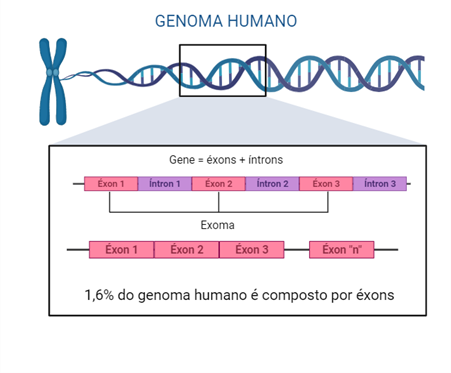
O exoma é a avaliação da parte do genoma composta pelos éxons, que são as regiões dos genes responsáveis pela codificação das proteínas. Uma característica marcante é que os éxons representam apenas uma pequena fração do código genético, estimada em cerca de 1,6% no genoma humano. No entanto, eles contêm a maior parte das variantes genéticas (popularmente chamadas de “mutações”) conhecidas com impacto direto nas características clínicas e no funcionamento do organismo.
Figura 1: Representação da estrutura dos genes humanos. O gene é composto de éxons, retratados em rosa, e íntrons, retratados em lilás. O sequenciamento do exoma avalia apenas as partes em rosa do gene, ou seja, os éxons.
Qual a relação entre os tipos de doenças e a frequência das variantes genéticas?
As variantes genéticas são mudanças que ocorrem na sequência do DNA de um indivíduo. Podem ser classificadas de acordo com seu impacto clínico ou pelo tipo de alteração que ocorre.
As variantes comuns contribuem para a diversidade de características humanas, incluindo aspectos cognitivos e comportamentais. Podem ser herdadas dos pais e, geralmente, possuem baixo impacto clínico quando consideradas individualmente. Porém, quando combinadas, essas variantes podem conferir risco aumentado para a manifestação de doenças comuns, como diabetes, pressão alta e depressão.
Por outro lado, em síndromes genéticas raras, frequentemente, uma única variante isolada é suficiente para ser causal. Mutações raras têm baixa frequência populacional e, em uma dada família, geralmente ocorrem pela primeira vez na formação dos gametas de um dos pais da pessoa com a síndrome.
O que são as variantes de nucleotídeo único?
Dentre os vários tipos de alterações genéticas, temos as “variantes de nucleotídeo único” (SNVs, do inglês, single nucleotide variants), que são caracterizadas por afetarem poucos pares de bases do código do DNA. A associação de SNVs com doenças depende, principalmente, da localização no genoma. SNVs podem ser raras ou comuns variando de acordo com o local em que ocorrem. Muitas SNVs acontecem em regiões não codificantes. Logo, parte dessas mutações podem não causar alterações significativas no organismo.
Para que serve o exoma?
O sequenciamento do exoma é uma ferramenta de diagnóstico preciso, com um direcionamento assertivo, nos casos de SNVs em regiões codificadoras de proteínas, ou seja, exons.
O sequenciamento do exoma é pincipalmente recomendado em casos de pacientes com suspeita de síndromes genéticas raras que envolvem o transtorno do espectro autista, encefalopatias epilépticas, atraso do desenvolvimento e/ou deficiência intelectual. Esse exame é frequentemente usado em crianças com alterações do neurodesenvolvimento em que os pais não apresentam a mesma característica.
Pessoas que possuem histórico familiar sugestivo de doença hereditária, sem diagnóstico conclusivo, também podem ter indicação para a avaliação do exoma. Devido ao seu alto custo, ele pode ser realizado depois de outros testes genéticos clássicos, como cariótipo, terem retornado resultados negativos ou inconclusivos.
Quando o exame do exoma não é a melhor escolha?
Quando um segmento genômico maior, envolvendo uma fração substancial de um gene, um gene inteiro ou até mesmo diversos genes, é deletado, duplicado, invertido, ou translocado, a avaliação por sequenciamento de exoma não é o método de escolha para a sua detecção. Além disso, a avaliação do exoma não será efetiva para a detecção de variantes localizadas fora das regiões codificadoras dos genes.
As alterações genéticas são frequentemente associadas com doenças ou síndromes. Existem diversos tipos dessas alterações, sendo a quantidade de material genômico envolvido um dos fatores por trás de seu impacto clínico. Dentre eles, os dois tipos principais são “variantes de nucleotídeo único” e “alterações cromossômicas”.
As “variantes de nucleotídeo único” são caracterizadas por afetarem poucos pares de bases (ou “letras”) do código do DNA, de forma que a sua associação com doenças depende, principalmente, da localização no genoma. Já as alterações cromossômicas geralmente atingem milhões de pares de bases de DNA. Quando uma grande quantidade de material genético é alterada, aumenta-se o risco de ocorrência de síndromes.
Como são classificadas as alterações cromossômicas?
Essas alterações podem ser classificadas como numéricas ou estruturais. Quando o genoma do indivíduo apresenta uma quantidade de cromossomos inteiros em excesso ou faltando, chamamos essa variação de “numérica”. O exemplo mais conhecido de alteração cromossômica numérica é a síndrome de Down, que é causada pela trissomia do cromossomo 21, ou seja, um cromossomo 21 a mais no genoma.
Por outro lado, as alterações cromossômicas estruturais acontecem quando pedaços (ou “segmentos”) de cromossomos estão a mais ou a menos, ou ainda, quando eles mudam de lugar. As alterações cromossômicas estruturais mais frequentes implicam em duplicações ou deleções de grandes segmentos genômicos, que geralmente envolvem diversos genes. Para que essas alterações ocorram, acontecem quebras no segmento de DNA que compõe o cromossomo. Qualquer cromossomo pode sofrê-las, havendo grande heterogeneidade de tamanhos de segmentos genômicos afetados e, consequentemente, de alterações clínicas associadas.
Tipos de avaliação de alterações cromossômicas
Os exames de análises cromossômicas geralmente são solicitados quando o indivíduo possui uma malformação congênita associada a alterações no neurodesenvolvimento, sem que haja suspeita de uma síndrome genética causada por variantes de nucleotídeo único. Outros casos em que a avaliação de alterações cromossômicas pode auxiliar no diagnóstico são atraso da puberdade, baixa estatura, infertilidade ou abortos de repetição.
O estudo dos cromossomos surgiu com a citogenética clássica, que se utiliza do exame do cariótipo por bandamento G. Apesar de ser comumente utilizado como primeira triagem diagnóstica para alterações cromossômicas, o exame apresenta uma baixa resolução. Por isso, ele impede a determinação precisa da localização de quebras cromossômicas ou o tamanho do segmento genômico afetado pelas alterações estruturais.
Nos últimos anos, o estudo de alterações cromossômicas estruturais com metodologias moleculares de alta resolução tem permitido o reconhecimento de genes e de regiões genômicas responsáveis por determinadas características humanas. Em muitos casos, após a identificação do segmento cromossômico duplicado ou deletado, é possível oferecer um melhor prognóstico e aconselhamento genético às pessoas com alterações cromossômicas estruturais e suas famílias.
Pelo fato de as alterações estruturais serem mais complexas e heterogêneas, elas são estudadas com maior frequência na pesquisa científica. Esses estudos se propõem a correlacionar o segmento cromossômico afetado pela alteração estrutural com o quadro clínico do indivíduo, o que é chamado de “correlação genótipo-fenótipo”. Para que essa correlação aconteça, os pontos de quebra dos cromossomos precisam ser definidos com alta resolução por metodologias moleculares.
Testes de alta resolução para avaliação de pontos de quebra cromossômica
Para melhor delineamento molecular e identificação das alterações cromossômicas, diversas técnicas foram desenvolvidas e aprimoradas. A principal delas é o array genômico, uma técnica de alta resolução que analisa o genoma em sua totalidade, incluindo detecção de ganhos e perdas no material genético.
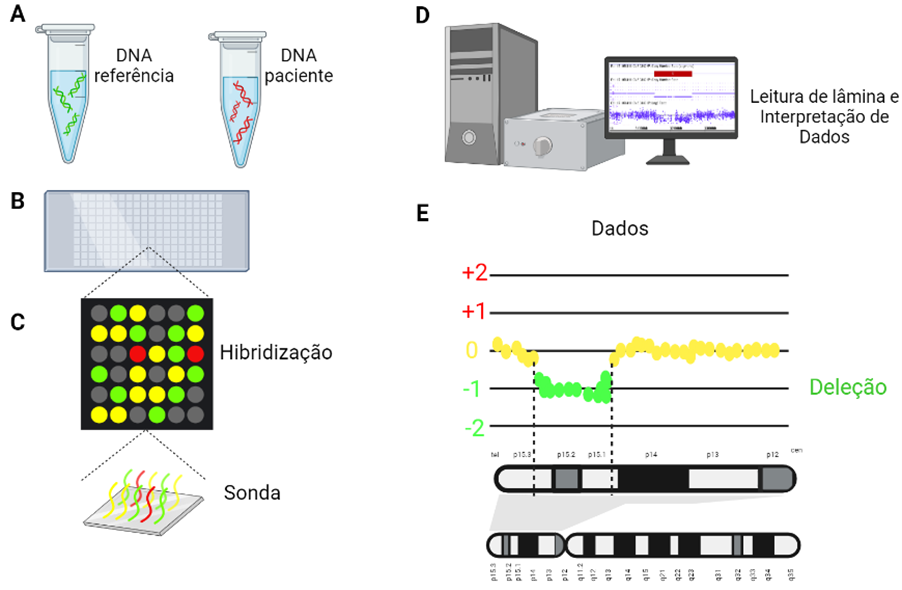
Existem alguns tipos de array genômico, como por exemplo o array CGH (do inglês, Comparative Genomic Array), demonstrado na imagem abaixo.
O procedimento do array CGH requer duas amostras de DNA, uma amostra de referência (ou seja, material genético de uma pessoa sem alterações cromossômicas) e uma amostra do paciente. Essas amostras de DNA são geralmente obtidas a partir de uma coleta de sangue. Os fragmentos genômicos são marcados com fluorescência, neste caso, verde para DNA referência e vermelho para a amostra do paciente.
Em uma lâmina, há a disposição de milhares de segmentos pequenos de DNA, contendo sequências complementares a regiões do genoma, chamadas de “sondas”. Essas sondas servem para avaliar a quantidade de material genético dos seus alvos genômicos, determinando se há perdas ou ganhos de DNA no genoma do paciente.
Como demonstrado na imagem abaixo, nas regiões em que o número de cópias no DNA da amostra teste (do paciente) for igual ao número de cópias da amostra de referência, o verde e vermelho são misturados de forma equivalente, e as sondas ficarão amarelas. Já nas regiões em que houver uma deleção no paciente, as sondas ficarão verdes, pois a fluorescência do DNA de referência vai se sobressair. Se houver duplicação no paciente, as sondas ficarão vermelhas.
A avaliação da emissão de fluorescência ocorre depois que os segmentos de DNA provenientes das amostras de material genético se ligarem às sondas. A lâmina de array é então analisada por um software, que quantifica a fluorescência emitida por cada sonda e a traduz em número de cópias de DNA do genoma do paciente.
Procedimento do array CGH. (A) Amostras de DNA de referência e do paciente. (B) Lâmina com milhares de sondas. (C) Avaliação sobre quantidade de material genético dos alvos das sondas no genoma, determinando se há perdas ou ganhos de DNA na amostra do paciente. (D) Avaliação da emissão de fluorescência por um software de análise de dados. (E) Tradução do sinal de fluorescência emitida por cada sonda em número de cópias de DNA no genoma do paciente.
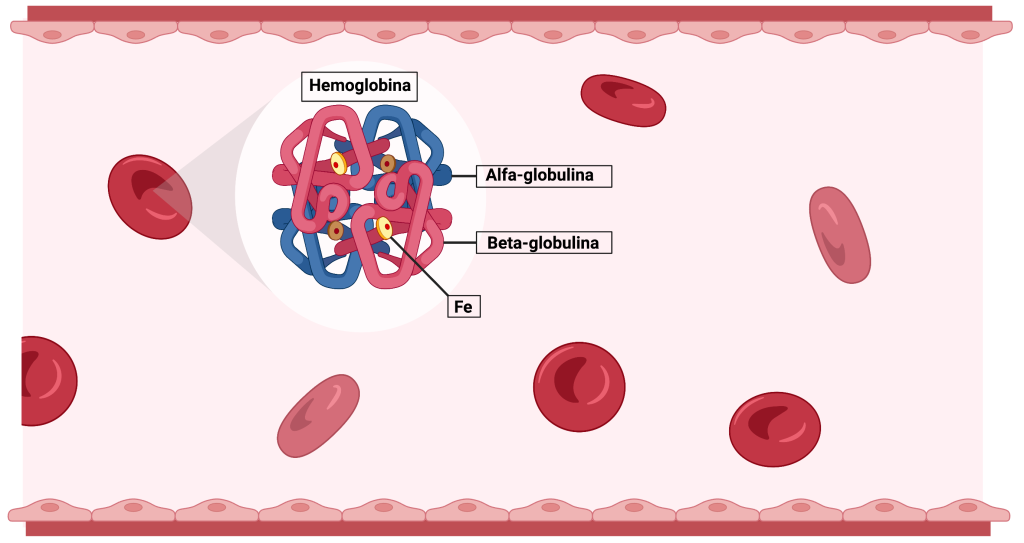
As talassemias são doenças sanguíneas de etiologia genética que fazem com que os pacientes produzam menos glóbulos vermelhos saudáveis e menos hemoglobina que o normal. Os dois principais tipos de talassemia são Alfa e Beta e base genética das talassemias envolve diferentes genes. Os genes associados às talassemias codificam globulinas e fazem com que a hemoglobina apresente quatro cadeias de proteínas (duas alfa-globinas e duas beta-globinas).
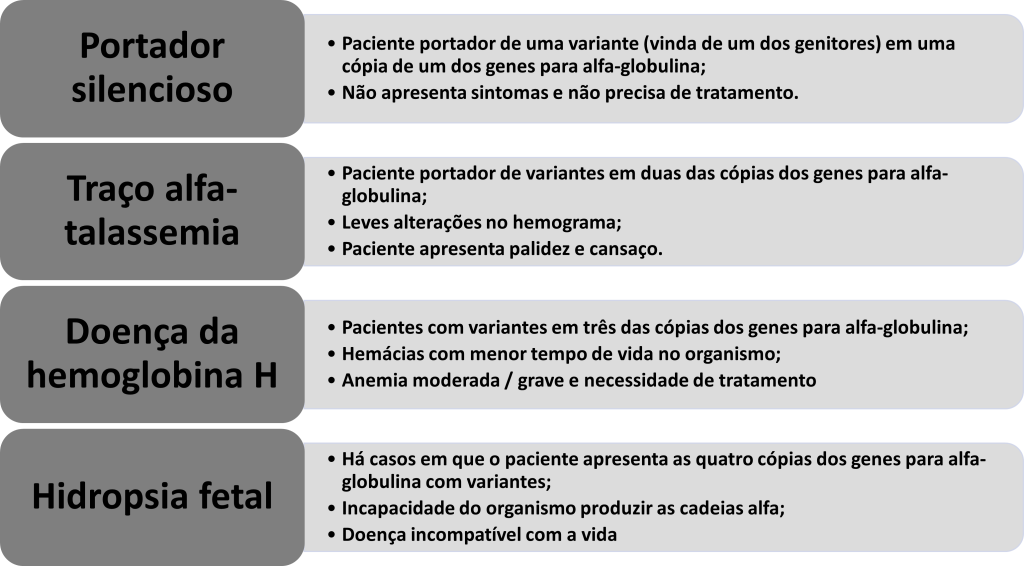
Para a produção de globina alfa são necessários um total de 4 cópias dos genes (ou seja, dois genes, sendo duas cópias de cada gene) que codificam a globulina alfa funcionantes. Esses genes estão localizados no cromossomo 16, estando dois alelos em cada cromossomo (dois no cromossomo 16 materno e dois no paterno). Os pacientes que apresentam talassemia alfa têm mutações nestas cópias de genes e podem apresentar 4 formas da doença:
A talassemia beta, por sua vez, surge com um defeito em uma cópia de gene do cromossomo 11, afetando a produção de globina beta. Este é o tipo mais visto em pacientes com talassemia no mundo e pode ser classificada como:
O diagnóstico das talassemias é geralmente feito por meio de exames de sangue, que incluem hemograma completo com análise de lâminas (os pacientes com talassemia têm menos glóbulos vermelhos saudáveis e menos hemoglobina) e exames especiais de hemoglobina (pacientes com talassemia apresentam problemas nas cadeias alfa e beta globina da hemoglobina). Além disso, a quantidade de ferro no sangue pode ser um sinal de alerta para o diagnóstico. O paciente com talassemia deve seguir seu plano de acompanhamento, fazendo hemogramas completos mensais e testes para níveis de ferro no sangue a cada três meses; testes anuais para função cardíaca, função hepática, infecções virais (hepatite B, C e HIV), investigação do acúmulo de ferro no fígado; testes anuais de visão e audição; exames regulares para garantir o funcionamento das transfusões de sangue; exame de imagem para acompanhar o tamanho do baço. Além disso, por ser uma doença hereditária, estudos genéticos também são importantes para ajudar no diagnóstico preciso da doença. Estes estudos envolvem a junção de um histórico médico familiar com a realização de exames em parentes a partir da coleta de sangue. Os resultados genéticos podem mostrar se algum membro da família tem genes importante para a formação da hemoglobina alterados.
A Medicina Convencional utiliza uma abordagem reativa, isto é, tratando as doenças após elas já terem causado sintomas no paciente. Além disso, a medicina convencional é baseada em tratamentos generalizados e padronizados, que têm sua eficácia testada em escala populacional e que funcionam para a maioria das pessoas em um grupo grande de indivíduos.
Por outro lado, a Medicina de Precisão, com base em uma abordagem individualizada, é voltada para a prevenção de doenças e para a aplicação de terapias mais eficientes levando em consideração o perfil específico e as particularidades do organismo de um determinado indivíduo. Essa nova modalidade da medicina tende a proporcionar maior qualidade de vida aos pacientes, pois evita tratamentos mais longos e debilitantes. A medicina de precisão possibilita menores gastos públicos e privados com a saúde, já que é muito mais fácil e barato prevenir doenças do que tratá-las depois que os sintomas já apareceram.
A medicina de precisão é uma abordagem de acompanhamento ou tratamento personalizado ao paciente, com base, principalmente, no seu perfil genético e metabólico. Os recentes progressos em pesquisas na área de genética humana têm sido importantíssimos para a medicina de precisão. Após o Projeto Genoma Humano, que ocorreu de 1990 a 2003, houve um barateamento e uma maior disseminação de técnicas de genotipagem em larga escala, ou seja, técnicas que fazem a leitura de grande parte do código genético do indivíduo. A partir desses estudos, houve grandes avanços nas descobertas de biomarcadores genéticos para muitas doenças e para respostas a diversos medicamentos. Estudar a genética humana também nos permite desenvolver novas técnicas de diagnóstico e terapias. Recentemente, a descoberta do sistema de edição genética por CRISPR e o uso de células-tronco em terapias celulares abriu uma gama de possíveis aplicações para a medicina de predição.
A medicina de precisão pode ser aplicada em diversas áreas e para finalidades diferentes. predição de riscos de doenças, farmacogenética e auto-transplantes.
Predição de riscos de doenças:
A medicina de precisão leva em conta as variantes presentes no DNA de cada indivíduo para predizer se essa pessoa possui um risco genético maior de desenvolver certos problemas de saúde, como doenças cardiovasculares, câncer, doenças psiquiátricas, entre outros. Atualmente, muitas pesquisas dessa área estão focadas na descoberta de novas variantes genéticas (ou seja, “mutações”) que estejam associadas com doenças e na avaliação das consequências dessas variantes para a saúde dos indivíduos.
Os biomarcadores genéticos têm um importante papel na predição de doenças e tratamentos personalizados. Tratam-se de alterações no genoma que sinalizam a ocorrência ou a maior chance de ocorrência de processos relacionados a doenças. Os biomarcadores genéticos podem ser variações na sequência de DNA ou modificações epigenéticas, que alteram a expressão, a regulação ou o funcionamento dos genes. Por meio deles, é possível predizer o risco de um indivíduo desenvolver doenças no futuro ou como um paciente poderá responder a um determinado tratamento. A predição dos riscos genéticos de se desenvolver doenças é muito útil para que as pessoas obtenham um diagnóstico precoce e preciso, possibilitando, em alguns casos, a prevenção da enfermidade ou a escolha de um tratamento mais assertivo e com menos efeitos indesejados.
Farmacogenética
A Farmacogenética é a área que estuda como as características genéticas de cada pessoa podem influenciar na resposta de seu organismo a um determinado tratamento medicamentoso, como a eficácia e a duração do efeito de um medicamento, ou até mesmo seus possíveis efeitos adversos.
Dessa forma, o objetivo da farmacogenética é possibilitar a oferta de tratamentos personalizados de acordo com as características genéticas de cada indivíduo, de forma a minimizar efeitos colaterais e potencializar o efeito terapêutico.
Autotransplante
Ocorre quando tecidos, órgãos, células ou até mesmo proteínas são transportados de uma parte do corpo de uma pessoa para outra parte do corpo deste mesmo indivíduo (para saber mais sobre transplante, acesse a Edição 46 do Gibi Dona Ciência sobre Imunodeficiências).
O autotransplante, quando possível, geralmente é uma alternativa interessante em relação ao transplante alogênico (ou seja, quando o tecido, órgão ou célula recebidos provêm de outro indivíduo), pois há menores riscos que o paciente tenha rejeição ao transplante.
A possibilidade de se fazer terapias com células-tronco trouxe novas perspectivas para o uso dos autotransplantes. As células-tronco podem ser definidas como células indiferenciadas ou não especializadas. Elas não somente são capazes de originar outras células-tronco, como também se diferenciam em diversos outros tipos de células especializadas. A partir deste processo de diferenciação celular, as células-tronco podem ser utilizadas para restaurar a função de tecidos com danos ou criar novos tecidos in vitro (ou seja, fora de um organismo vivo).
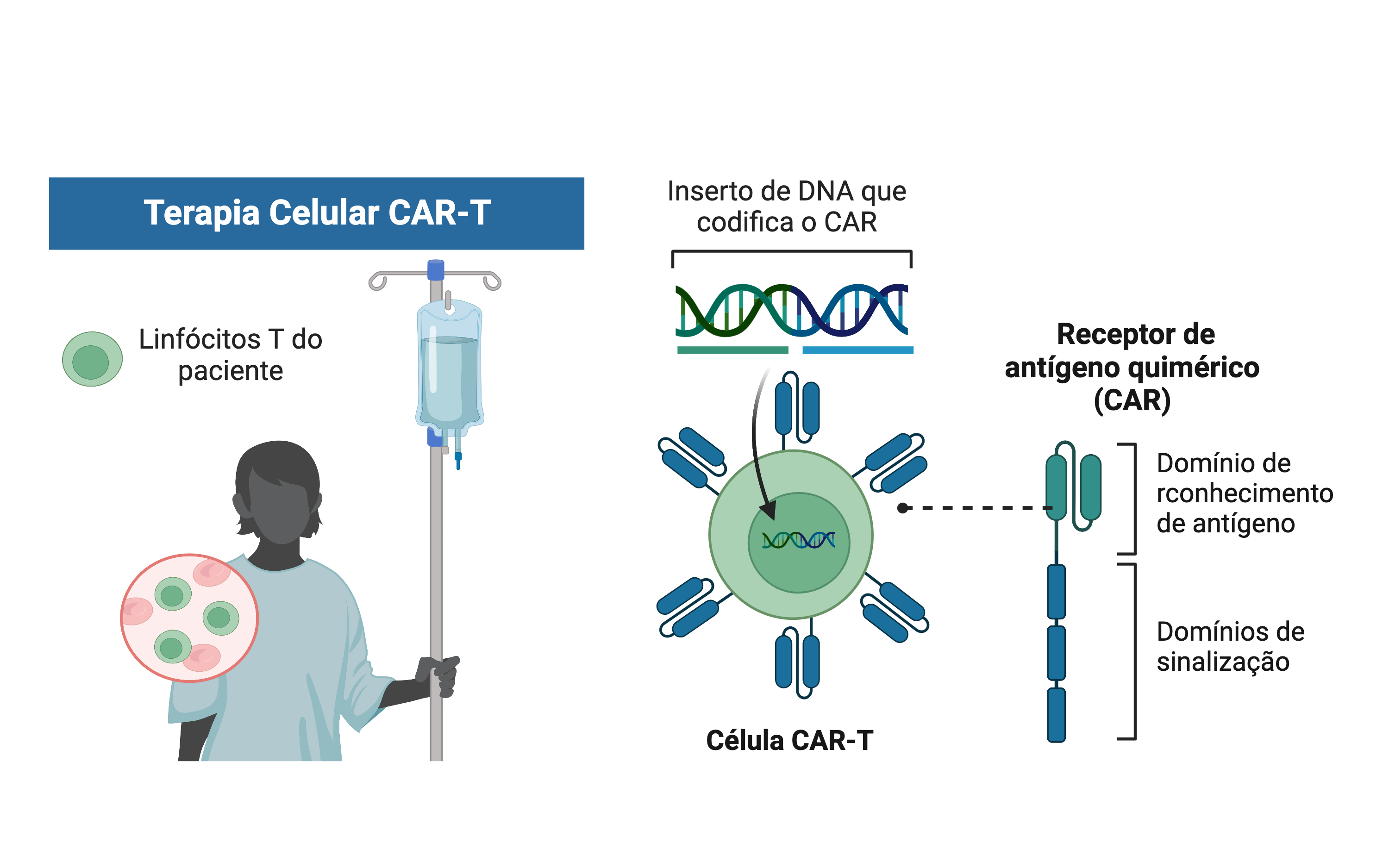
Um dos maiores exemplos de como o CRISPR pode revolucionar a área de saúde é o seu uso em terapia gênica para tratamentos contra o câncer. Uma dessas terapias baseia-se na produção de células CAR-T (do inglês, chimeric antigen receptor T-cell) a partir dos linfócitos T, que são células do sistema imune muito importantes para a defesa do nosso organismo contra agentes desconhecidos. Portanto, as células CAR-T são linfócitos T modificados geneticamente pela técnica de CRISPR e programados para reconhecer e combater células do câncer. Para a realização dessa terapia, os linfócitos T do próprio paciente são transformados em células CAR-T e inseridos novamente em sua circulação sanguínea. Ou seja, as próprias células do paciente são reprogramadas geneticamente para conseguir combater o câncer.
As perspectivas futuras da medicina de precisão estão relacionadas com a predição cada vez mais ampla e específica de doenças e com o crescimento da prevenção e do tratamento personalizados de problemas de saúde em um cenário onde procedimentos inovadores possibilitarão tratar enfermidades com grande impacto na mortalidade da população e que, até então, não possuíam cura.
Seja nosso cliente
Logística rápida, segura e rastreável, alto padrão tecnológico, equipes especializadas e consultoria personalizada: o parceiro ideal para o seu negócio.